Resource
Single cell/Spatial omics-related tools
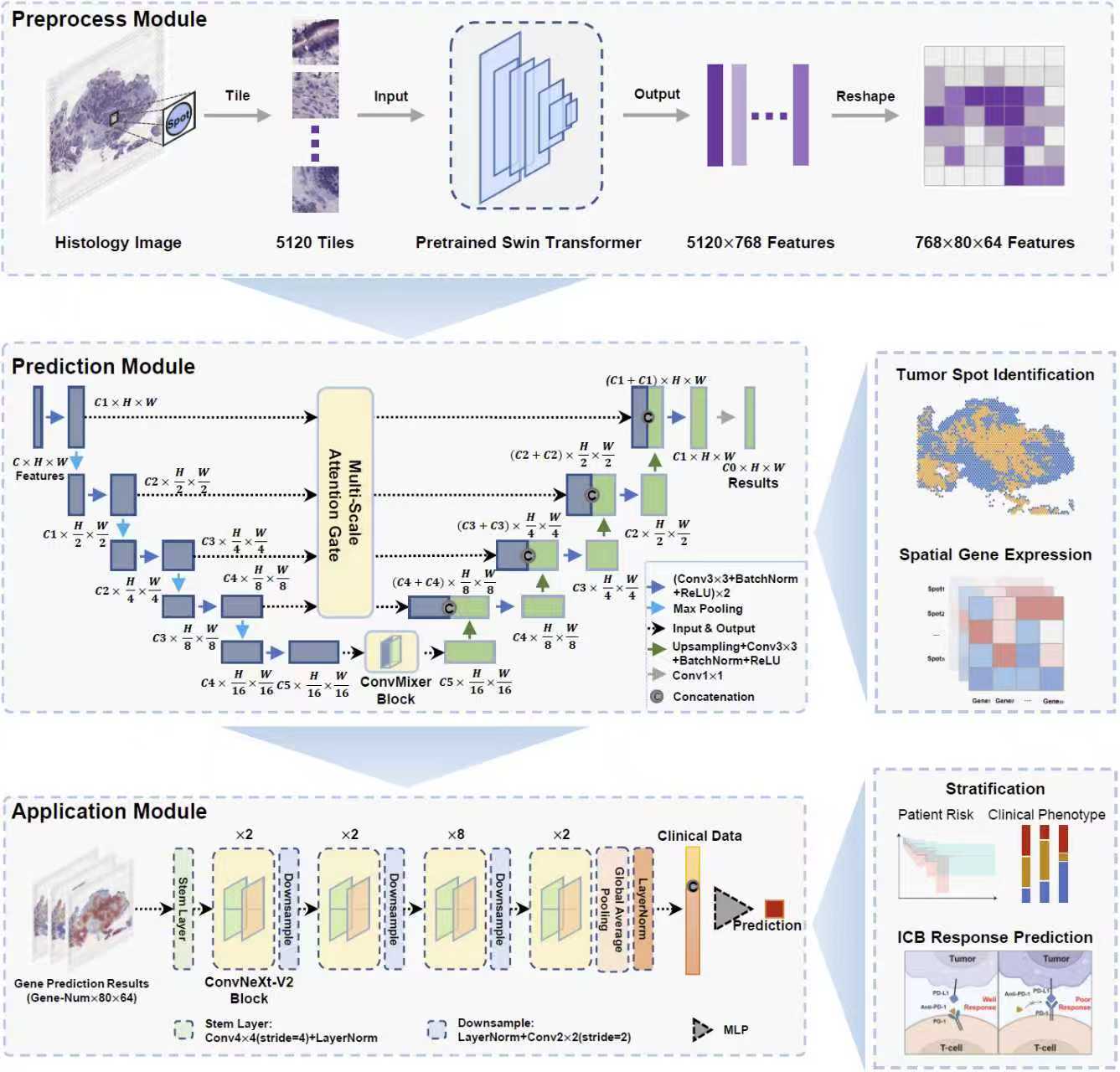
HiST
(https://github.com/Yelab2020/HiST)
HiST is a multi-scale convolutional deep learning framework for analyzing spatially resolved gene expression profiles (GEPs) and histological morphology. The tool accurately predicts tumor regions (e.g., breast cancer AUC: 0.96) and reconstructs spatial GEPs with high correlation (average PCC: 0.74). HiST also enables prognostic prediction (e.g., breast cancer C-index: 0.78) and immunotherapy outcome assessment, while revealing key regulatory networks.
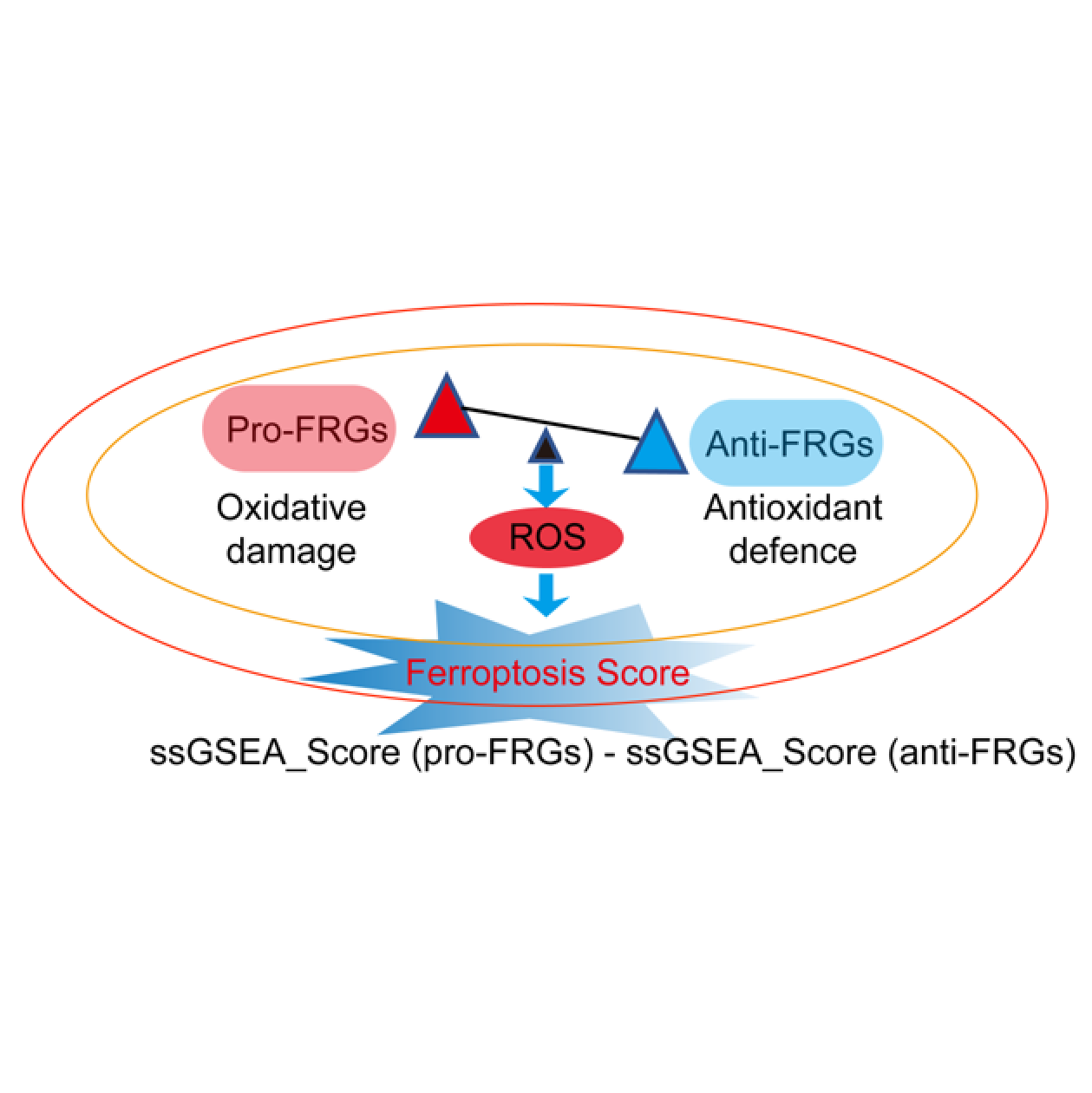
FPSOmics
(https://github.com/Yelab2020/FPSOmics)
FPSOmics is an R package to calculate ferroptosis score (FPS) and multidimensional ferroptosis-associated molecular signatures. FPSOmics designed a novel score model based on ferroptosis-related genes(FRGs) using single sample gene set enrichment analysis (ssGSEA). This package also include a propensity score matching (PSM) algorithm, which appropriately control the effects of clinical confounding factors, to calculate ferroptosis-associated molecular signatures.

Cottrazm
(https://github.com/Yelab2020/Cottrazm)
Cottrazm (Construct Tumor Transition Zone (Boundary) Microenvironment based on spatial transcriptomics) aims to construct the microenvironment of tumor boundary based on spatial transcriptomics, single-cell transcriptomics and HE-stained histological images. It consists of three core functions: determining the tumor boundary (Cottrazm-BoundaryDefine), deconvoluting spatial transcriptomics (Cottrazm-SpatialDecon), and reconstructing a spatial gene expression matrix for sub-spots (Cottrazm-SpatialRecon).
Taken together, Cottrazm provides an integrated tool framework to dissect the tumor spatial microenvironment and facilitates the discovery of functional biological insights, thereby identifying therapeutic targets in oncologic ST datasets.
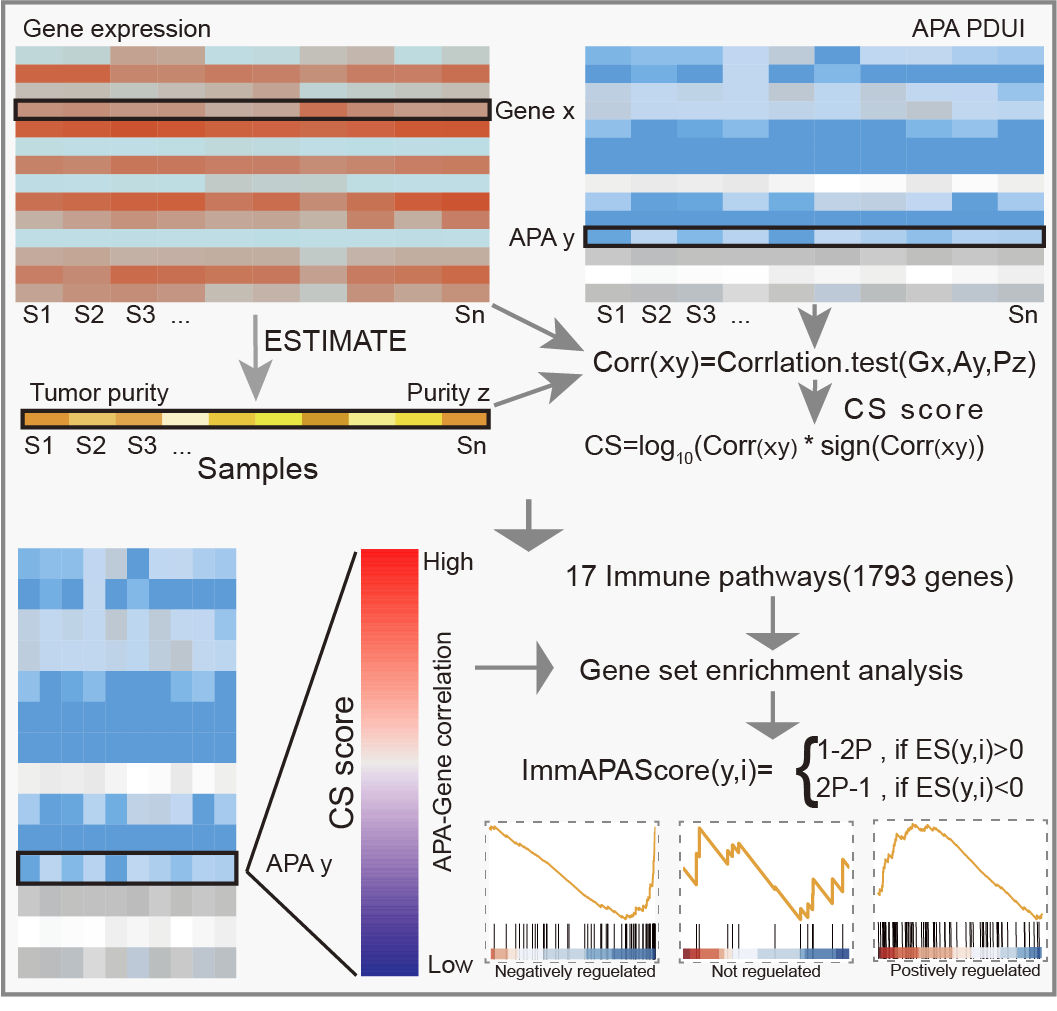
ImmAPAScore
(https://github.com/Yelab2020/ImmAPAScore)
ImmAPAScore (Immune-related Alternative Polyadenylation Events Score) is an integrated algorithm estimating the relationship of APA events and tumor immunity to characterize the regulatory landscape of APA events in tumor. The pipeline including construction of ImmAPAScore to identify immune-related APA in human cancer, identification of tumor-immunity-related APA events, identification of top-ranked APAs, and construction of an ICB-related ImmAPA score model to predict immunotherapy efficacy.
Database
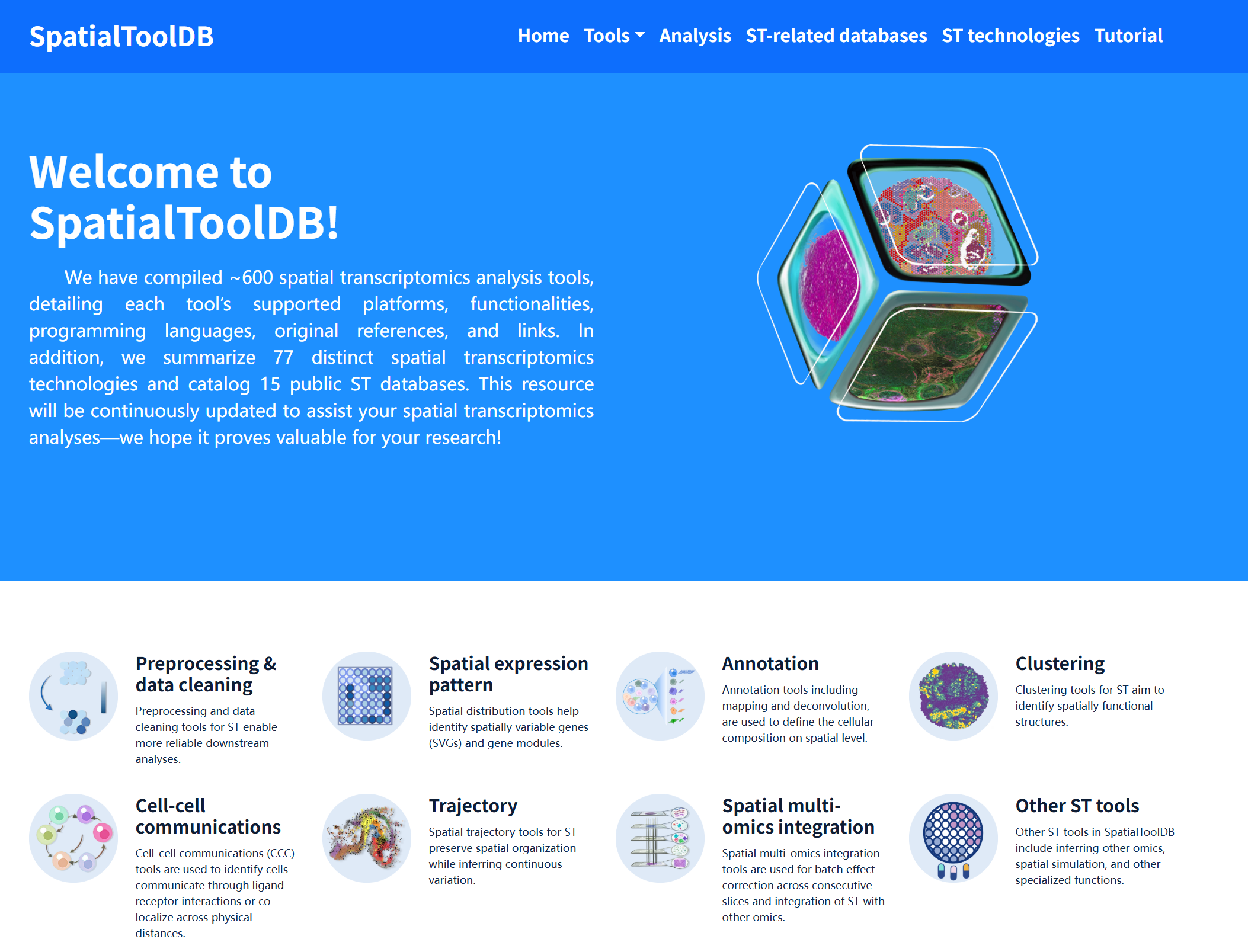
SpatialToolDB
(https://www.spatialtooldb.yelab.site/)
SpatialToolDB is a comprehensive resource designed to systematically catalog analytical methods and resources in the field of spatial transcriptomics. Here, we compiled approximately 600 spatial transcriptomics analysis tools, with detailed annotations including supported platforms, core functionalities, programming languages, original publications, and relevant links. In addition, we curated 77 distinct spatial transcriptomics technologies and summarized 15 publicly available spatial transcriptomics databases. SpatialToolDB is continuously updated to support method selection, benchmarking, and workflow design, providing a valuable reference for spatial transcriptomics research.
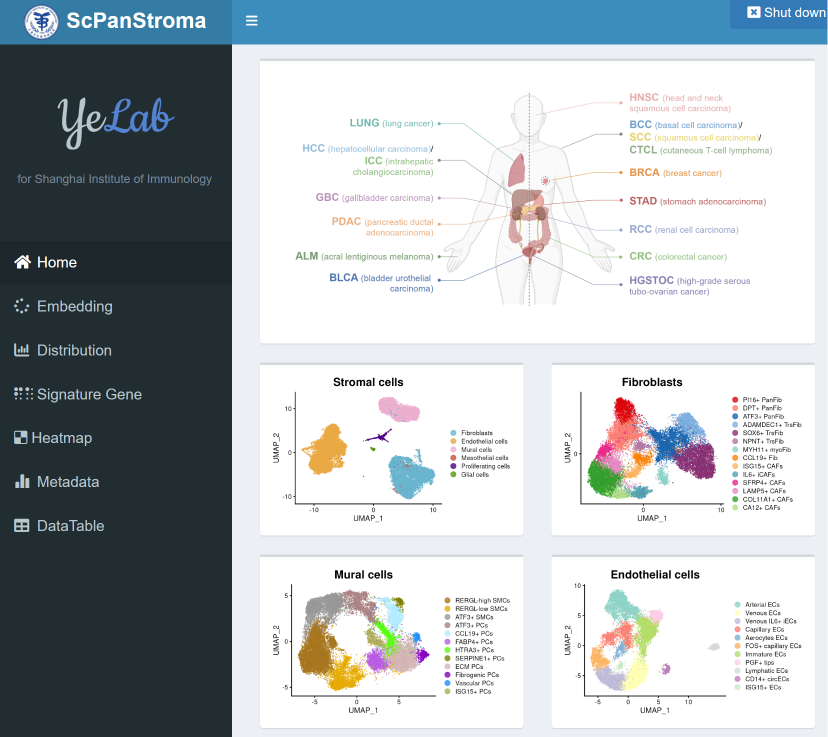
scPanStroma
(http://www.scpanstroma.yelab.site/)
scPanStroma (Single cell pan-cancer stroma) is an interactive web-based tools for exploring tumor-associated non-immune stromal cells in pan-cancer. We performed a pan-cancer analysis of 214,972 stromal cells using single-cell RNA-sequencing from 258 patients across 16 cancer types, and developed the interactive web-based tool. It includes modules of loading data/inquiring genes, generating tables and figures for analysis results and downloading our single-cell information.
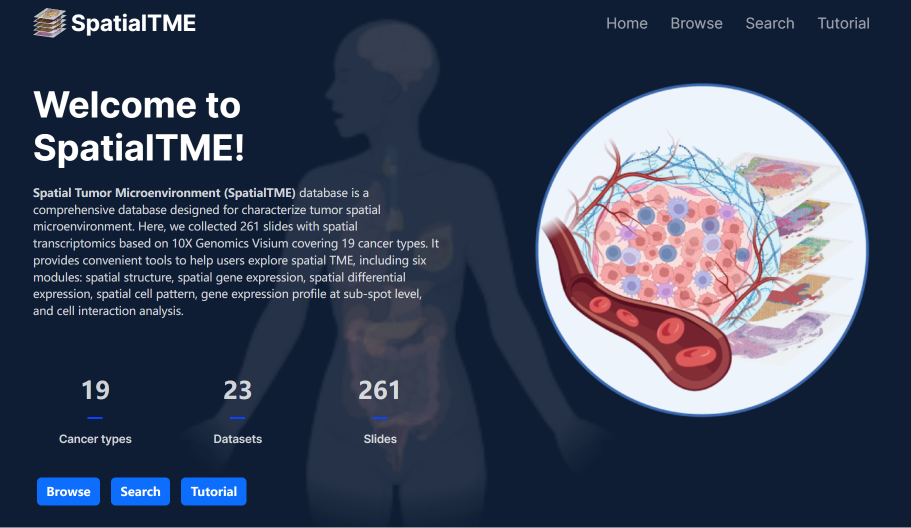
SpatialTME
(https://www.spatialtme.yelab.site/)
SpatialTME (Spatial Tumor MicroEnvironment) is a comprehensive database designed for characterize tumor spatial microenvironment. Here, we collected 261 slides with spatial transcriptomics based on 10X Genomics Visium covering 19 cancer types. It provides convenient tools to help users explore spatial TME, including six modules: spatial structure, spatial gene expression, spatial differential expression, spatial cell pattern, gene expression profile at sub-spot level, and cell interaction analysis.
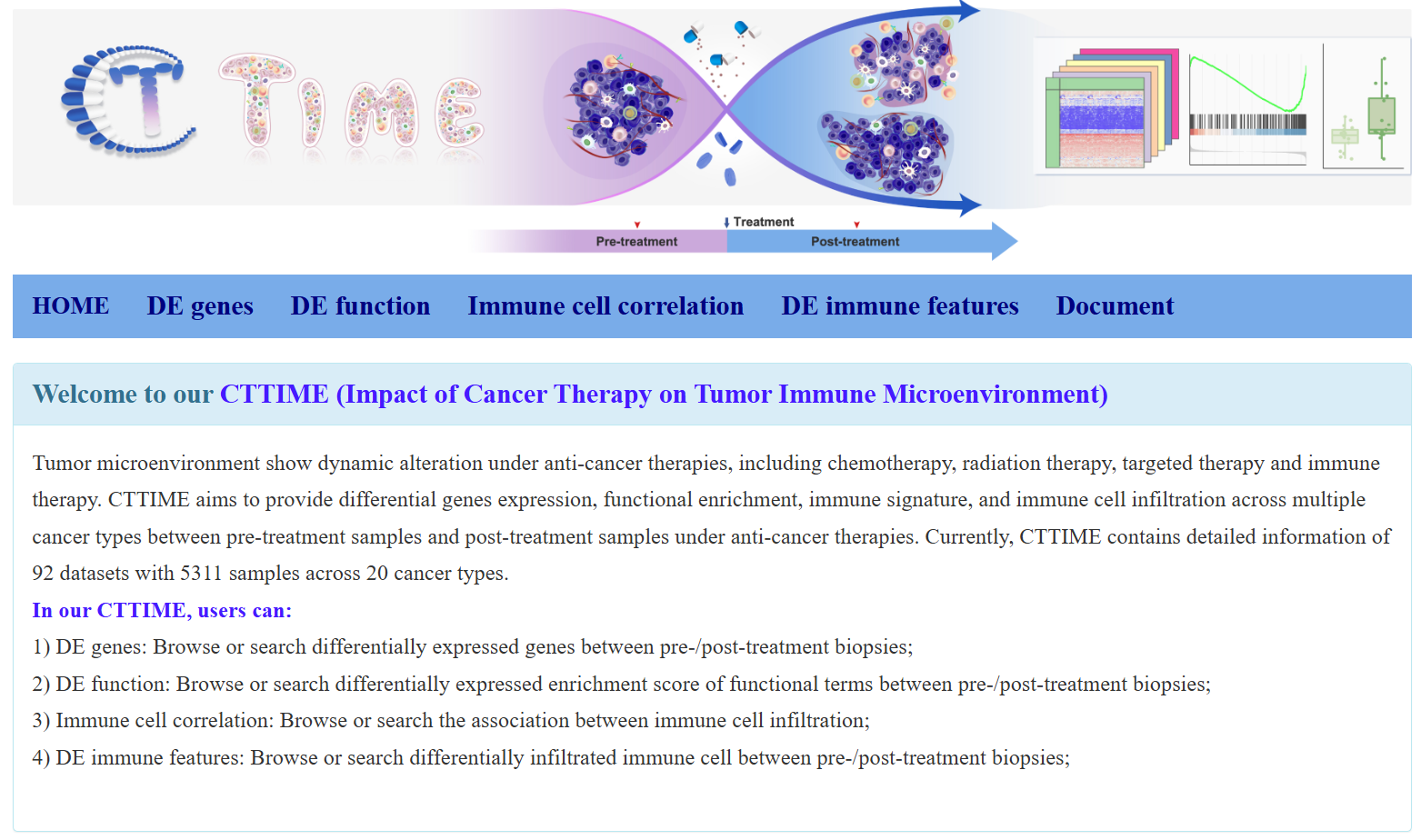
CTTIME
(http://www.cttime.yelab.site/)
CTTIME (Cancer Therapy’s Impact on Tumor Immune Microenvironment) is designed to identify dynamic molecular profiles under various anti-cancer therapies. This database comprised 92 transcriptomic datasets, including 5311 pre- and post-treatment biopsies from 20 cancer types and 9 treatment strategies. It includes four modules: DE gene, DE function, Immune cell correlation. DE immune features.